Review
Discovery and development of COVID-19 vaccines and therapeutics: nonclinical perspectives
2024 Volume 49 Issue 3 Pages 79-94
Details
2024 Volume 49 Issue 3 Pages 79-94
The development and regulatory review of BNT162b2, a COVID-19 vaccine, and PaxlovidTM (nirmatrelvir tablets/ritonavir tablets), a COVID-19 therapeutic, are benchmarks for accelerated innovation during a global pandemic. Rapid choice of the SARS-CoV-2 spike protein and main protease (Mpro) as targets for the vaccine and therapeutic, respectively, leveraged the available knowledge of the biology of SARS-CoV-2 and related viruses. The nonclinical immunogenicity and safety of BNT162b2 was rigorously assessed. Likewise, a comprehensive nonclinical safety assessment was conducted for the therapeutic candidates, lufotrelvir (PF-07304814) and nirmatrelvir (PF-07321332). The development and regulatory review of BNT162b2 and Paxlovid was enabled through close collaboration of the pharmaceutical industry with regulatory agencies and public health organizations. This experience highlights approaches that could be adopted for pandemic preparedness including risk-based investment strategies, conduct of activities in parallel that normally are conducted sequentially, quick kill decisions, simultaneous evaluation of multiple candidates, and use of flexible, established vaccine platforms.
Pharmaceutical research and development (R&D) have significantly contributed to human health, disease eradication, and lifespan extension over the past several decades. The global response to the COVID-19 pandemic, which led to the rapid development and deployment of various vaccines and therapeutics, is a prime example. This swift action had profound implications, not only for individual health and livelihoods, but also for the global economy. Furthermore, the past two decades have witnessed a doubling in the number of drugs under pharmaceutical R&D, with a significant emphasis on therapeutic areas of unmet medical need, particularly oncology and rare diseases.
The path to regulatory approvalIn developing new drugs, only a small percentage of compounds ultimately receive regulatory approval. This journey typically takes around ten years and, in some cases, begins with a pool of approximately 20,000 compounds in the discovery stages. The most significant and expensive drop-off in the clinical stage occurs during Phase 2, mainly due to efficacy considerations. While compound attrition is higher for small molecules versus biological modalities; once a drug enters clinical development, the success rate is similar for small and large molecules. In the past decade, there has been a decrease in terminations during Phase 3, primarily because higher quality substrates are being advanced to this stage. Differentiating comparative safety remains a critical factor to consider during Phase 3.
The role of nonclinical safety in drug developmentDrug discovery necessitates extensive collaboration among subject matter experts from various disciplines. This ensures the selection of the right targets and molecules, and clinical development is conducted in the appropriate patient population. Nonclinical safety is a focal point in this process.
Novel modalities, such as cell and gene therapy and ribonucleic acid (RNA) therapeutics, necessitate innovative safety approaches compared to small molecules and traditional biologics. However, the primary objectives for these modalities remain consistent: identifying target organs of toxicity, determining dose response, therapeutic index, reversibility, and monitorability. This information guides the selection of the initial clinical dose and provides further guidance as the candidate progresses through clinical trials.
Translational safety data from over 180 compounds and molecules, as reported by Monticello et al. (2017), demonstrates a specificity of over 80% concordance between nonclinical and clinical data, with beagle dogs being the most accurate predictors of negative outcomes in humans. However, the sensitivity is around 50%, with non-human primates (NHPs) being the strongest predictors. When the same target organ is identified in both species, the positive predictive value (PPV) increases. Across test species and target organs, negative predictive value (NPV) was the stronger predictive performance measure. Thus, a lack of toxicity in animal studies strongly predicts a similar outcome in humans in the clinic.
Nonclinical safety data for biologics have demonstrated a robust translation of human adverse drug reactions (ADRs) (Pfizer internal unpublished data). This means that the safety signals identified in nonclinical, non-human studies often accurately predict the adverse reactions observed in humans during clinical trials. This strong correlation is crucial as it allows for the early identification and mitigation of potential risks before a biologic is administered to humans.
Decades of experience in pharmaceutical R&D have equipped us with the knowledge to select the right targets, modalities, and patient populations for clinical trials. Specifically, in the field of toxicology, we have learned to conduct the appropriate safety studies that predict human safety signals; however, bringing a compound to the market still often takes approximately a decade. Therefore, under special circumstances, such as the COVID-19 pandemic, innovative lightspeed approaches are necessary to tackle emergencies.
The advent of the COVID-19 pandemic posed a significant challenge to the global scientific community. The established drug development cycle, which typically spans several years, was suddenly deemed too lengthy in the face of a rapidly spreading virus. However, the collective experience and knowledge amassed over decades of R&D provided a beacon of hope and a foundation to build accelerated strategies.
COVID-19, a disease with a wide spectrum of manifestations, ranging from the death of infected cells to the release of proinflammatory cytokines leading to multiorgan failures and death (Toussi et al., 2023), necessitated an urgent response.
Global response to COVID-19: vaccine and therapeutic developmentThe dire need for effective therapies and vaccines was underscored by the rapid escalation of the pandemic, with daily global deaths peaking at nearly 10,000 in the early stages. This urgency catalyzed worldwide efforts to find COVID-19 vaccines and therapies. By August 2022, the global response was evident, with approximately 10 vaccines approved and over 20 authorized in countries worldwide. These include vaccines based on various platforms, such as inactivated viruses, protein subunits, DNA plasmids, adenoviral vectors, and mRNA (Table 1).
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/49_79-t1.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09) mRNA vaccine platform and safety
mRNA vaccine platform and safety
Current mRNA vaccines, a relatively new modality, are composed of two basic components: mRNA and a lipid nanoparticle (LNP). The mRNA segment includes a 5’ cap, 5’ and 3’ untranslated regions, the sequence for the antigen of interest, and a polyadenylated (PolyA) tail. Various modifications, such as the use of modified nucleosides (eg, pseudouridine or methylpseudouridine), codon optimization, increased guanine-cytosine (GC) nucleotide content, and specific cap types, enhance the stability and translatability of the mRNA (Leppek et al., 2022; Kim et al., 2022; Karikó et al., 2005; Kon et al., 2022) (Fig. 1).

RNA vaccine composition. RNA vaccines are composed of 1) RNA and 2) lipid nanoparticles (LNP). The RNA is comprised of a 5’cap, a 5’untranslated region (UTR), an open reading frame (ORF) which encodes the antigen of interest (may be a full protein or only part of a protein), a 3’ UTR, and a polyadenylated tail (AAAn). In addition, the RNA sequence is codon as well as guanidine and cytodine content are optimized. A modified nucleoside, such as 1-methylpseudouridine (Ψ), is also incorporated in order to decrease the innate immune response to the vaccine components. The LNP are typically composed of 4 lipids: an ionizable cationic lipid, a polyethylene glycol (PEG)-lipid, cholesterol, and a phospholipid. The RNA is mixed with the lipids in a process that encapsulates the RNA inside the LNP.
Vaccine LNPs generally consist of 4 components: an ionizable cationic lipid, polyethylene glycol (PEG) linked lipid, cholesterol, and a naturally occurring phospholipid (Reichmuth et al., 2016). These components facilitate the self-assembly of the LNPs into virus-sized (~100 nm) particles, promote the endosomal release of mRNA into the cytoplasm, increase the half-life of the formulations, stabilize the particles, and support the lipid bilayer structure. LNPs also enhance cellular uptake of the RNA vaccine, protect against nuclease degradation, and may contribute to innate immune responses at the injection site.
Despite the relative novelty of the mRNA modality to the public, data has been generated related to its safety. mRNA vaccines are noninfectious and nonintegrating, eliminating the risk of infection or insertional mutagenesis (Pardi et al., 2018; Zhang et al., 2019; Houseley and Tollervey, 2009). Normal cellular processes degrade the mRNA. Its in vivo half-life can be regulated through various modifications and delivery methods. The mRNA and subsequent protein expression are transient due to the relatively short-lived nature of mRNA. The inherent immunogenicity of the mRNA, attributed to RNA pathogen sensors such as toll-like receptors 7 and 8 (TLR 7/8), can be decreased through nucleoside modifications to enhance the safety profile further (Karikó et al., 2005; Kon et al., 2022). mRNA expression depends on the LNP distribution, with the lipids being metabolized and eliminated or eliminated intact. Intramuscular administration results in local protein expression primarily at the injection site and, to a lesser extent, in the liver.
Antigen selection for COVID-19 mRNA vaccinesThe strategic selection of the spike protein as the target antigen for COVID-19 vaccines was instrumental in the development process. The SARS-CoV-2 virus comprises 4 structural proteins: the spike glycoprotein, nucleocapsid protein, membrane protein, and envelope protein (Malone et al., 2022). The spike (S) protein, forming a trimer, creates the characteristic crowns or spikes on the virus’s surface, facilitating viral entry into the host cell by binding to the angiotensin-converting enzyme 2 (ACE2) on the host cell surface (Li and Boix, 2021; Zhou et al., 2020).
Other coronaviruses utilize different cellular receptors for host cell entry, such as dipeptidyl peptidase-4 (DPP-4) for Middle East Respiratory Syndrome (MERS) or aminopeptidase N (APN) for numerous alpha coronaviruses (Nassar et al., 2021). SARS-CoV-2 cell entry primarily occurs via two mechanisms: receptor-mediated endosomal host cell entry via ACE2 and viral membrane fusion with the host cell plasma or endosomal membrane, both initiated by the S protein binding to ACE2 (Jackson et al., 2022).
The S protein is pivotal in viral infectivity, serving as an accessible target on the virus’s exterior. Crucially, neutralizing antibodies generated against the spike glycoprotein can disrupt the interaction between the virus and its host cell receptor, inhibiting the virus’s entry into the cell and/or targeting cells expressing the spike antigen, including infected cells. Early in the COVID-19 vaccine development, this antigen choice was substantiated by research on SARS-CoV-1 and MERS, where vaccines targeting the S protein demonstrated protective effects, and patients infected with SARS-CoV-1 developed antibodies targeting the S protein.
Mechanism of action of mRNA vaccinesThe mRNA encoding the sequence for the antigen of choice is encapsulated into an LNP. In the case of BNT162b2, this is the S protein locked into a prefusion confirmation. After injection, the mRNA encapsulated into the LNP enters antigen-presenting cells, which are efficient at stimulating a broad immune response (Fig. 2).

Mode of action of RNA vaccines. 1) The RNA-LNP after intramuscular injection is taken up by cells at the injection site and in the draining lymph node. 2) The RNA-LNP travels through the endosomal pathway and the RNA is released into the cytosol. 3) As with any other cellular RNA, the host cell’s machinery translates the RNA and the encoded protein, in this case the SARS-CoV-2 Spike protein, is produced and processed. 4) Fragments of the encoded protein are presented by antigen-presenting cells via major histocompatibility complexes. This leads to activation of B and T cells, producing both humoral (neutralizing antibodies) and T cell responses (killing of SARS-CoV-2-infected cells), and eventually longer-term memory cells.
Once inside the cell, the LNP travels through the endosomal pathway and eventually releases its contents, the mRNA, into the cytosol. The host cell’s machinery then translates the mRNA, and the protein antigen is expressed. Once produced, the whole S protein and its smaller fragments are presented to the immune system that recognizes the protein as “foreign,” resulting in a stimulation of T helper cells or CD4 T cells that activate other T cells and antibody-producing B cells. The antibodies can bind to the S protein on SARS-CoV-2 and neutralize the virus, which means the virus is no longer capable of infecting a cell. Also produced are CD8 T cells that can eliminate virally-infected cells. An important feature of mRNA vaccines is that they stimulate effective B cell and T cell memory responses, which ensures longer-term protection from viral infection and disease (Fig. 2).
mRNA-LNP vaccines and modern molecular technologiesThe mRNA-LNP vaccine modality and the mechanism of action of mRNA vaccines are a testament to the potential of modern molecular technologies to contribute to medical breakthroughs. Over the last decade, there have been significant advances in RNA technology, which has led to clinical trials of mRNA vaccines for various prophylactic and therapeutic indications (Dolgin, 2021; Wallis et al., 2019). The rapid development of an effective vaccine against SARS-CoV-2 grew out of this collective experience, with the first vaccine clinical trial initiated within a few months of the SARS-CoV-2 sequence publication using mRNA expressing the S protein.
In comparison to traditional vaccines, mRNA vaccines, which are produced using a DNA template, are relatively simple to produce and can be rapidly scaled up and developed. The manufacturing process is sequence-independent and is primarily dictated by the length of the RNA, the nucleotide and capping chemistry, and the purification of the product. The mRNA components and manufacturing process can be standardized to allow the production of any encoded protein immunogen. This modality is particularly suitable for rapid response to virus strain changes or emerging infectious diseases, as a platform approach may be pursued based on strong nonclinical and clinical safety and biodistribution data and evidence of immunogenicity in nonclinical species of the expressed gene of interest.
Translating nonclinical safety investigations into clinical applications: a comprehensive examination of steps, considerations, and strategiesThe development of the BNT162b2 COVID-19 vaccine is a testament to the power of scientific collaboration and innovation. A critical aspect of this development process was the nonclinical safety investigations, which provided the foundation for the subsequent clinical translations. Nonclinical immunogenicity and efficacy of BNT162b2 were evaluated in multiple animal models, including mice and non-human primates (NHPs).
In NHPs (Vogel et al., 2021), an immunogenicity and SARS-CoV-2 challenge study was conducted (Table 2). Serum was collected from the animals at multiple time points before and after injection with BNT162b2 or saline. Peripheral blood mononuclear cells (PBMCs) were also obtained for immunophenotyping, including ELISpot assays for IFNγ and IL-4 to differentiate type 1 T helper cells (Th1) versus type 2 T helper cells (Th2) responses and flow cytometry for intracellular cytokine profiling. Following the development of vaccine response in BNT162b2-administered animals, both groups were challenged with SARS-CoV-2, and various in-life and postmortem assessments were conducted. The results demonstrated that BNT162b2 elicited a robust immune response after prime-boost vaccination. Neutralizing antibody titers were 8-18 times that of convalescent human plasma. The response was Th1-biased, with a high frequency of CD4+ T cells and circulating S-specific CD8+ T cells. Importantly, the vaccine protected the lower respiratory tract after the SARS-CoV-2 challenge.
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/49_79-t2.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
The final nonclinical safety package (Table 2) for the vaccine included toxicity data from 2 rat repeat-dose toxicity studies evaluating 2 dose levels (30 and 100 µg) of 2 variants of BNT162b2, as well as data from a combined fertility and reproductive toxicity study using the human clinical dose (30 µg). The 2 BNT162b2 variants only differed in the codon optimization of their RNA sequence but produced identical spike proteins (Rohde et al., 2023; Bowman et al., 2021). Biodistribution studies were conducted using an RNA-encoding luciferase in the same LNP as the vaccine. These studies showed the highest distribution at the injection site, with the most protein or LNP observed outside the injection site and the draining lymph node in the liver (CHMP, 2021).
In the repeat-dose toxicity studies, 3 intramuscular (IM) vaccine injections, at doses up to 100 µg, were given one week apart to male and female Wistar Han rats (15/sex/group) (Fig. 3a). After the 3rd dose, a subset of rats (5/sex/group) underwent a 3-week recovery. The vaccine was well tolerated, with most findings associated with an inflammatory or immune response (Rohde et al., 2023). Clinically, rats administered BNT162b2 exhibited transient body weight loss and body temperature increases during the first week after dosing. Local reactions (edema and/or erythema) at the injection site were observed in the BNT162b2 groups that increased in incidence and severity after the 2nd or 3rd administrations.
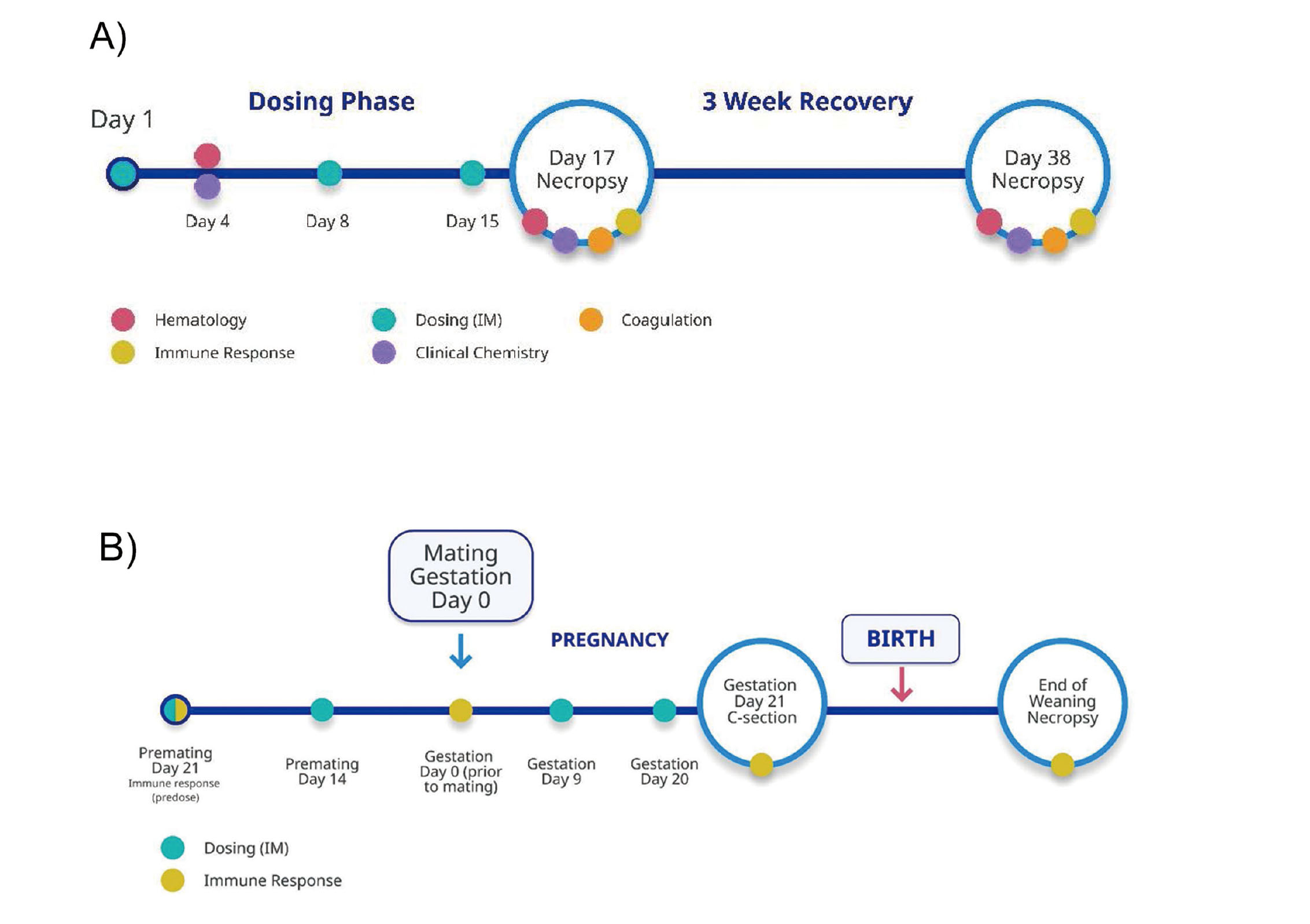
Study outlines for BNT162 repeat-dose and developmental and reproductive toxicity (DART) studies in rats. A) The two repeat-dose toxicity studies with BNT162 vaccine candidates consisted of a 17-day dosing phase (once weekly intramuscular dosing on Days 1, 8, and 15) followed by a 3-week recovery phase in male and female rats (15 rats/sex/group). Standard vaccine safety parameters were conducted including clinical observations, body weight, food consumption, assessments of body temperature, injection site dermal assessment, as well as assessments of clinical pathology parameters (Days 3, 17, and 38) as well as tissues from a subset of animals at each necropsy (10/sex/group at the end of the dosing phase and 5/sex/group at the end of the recovery phase. B) In the DART study with BNT162 vaccine candidates, vaccines were administered intramuscularly to female rats (44/group) twice prior to mating (21 and 14 days prior to cohabitation with untreated breeder males) and twice during gestation (Gestation Days 9 and 20). Half of the females underwent Caesarean (C)-section on Gestation Day 21. Uterine parameters, as well as fetal external, visceral, and skeletal exams were conducted. The remaining females were allowed to naturally deliver their pups and both were followed out through weaning (21 days after birth), including evaluations of offspring clinical observations, body weight, and some developmental milestones.
In-life findings resolved before the next vaccine administration or by the end of the 3-week recovery. Microscopically, vaccine-related findings included acute inflammation and edema at the injection site, increased cellularity (lymphocytes) in the lymph nodes and spleen, and increased hematopoiesis in the bone marrow and spleen. These microscopic findings were associated with clinical pathology observations of higher white blood cell counts and acute phase reactant concentrations, lower platelet and reticulocyte counts, and lower RBC parameters. In addition, minimal periportal hepatocellular vacuolation, without evidence of liver damage, was observed and attributed to LNP lipid uptake. All of the above-mentioned clinical pathology and microscopic findings were partial to fully resolved by the end of the 3-week recovery. Anti-spike neutralizing antibody responses were present in both males and females.
In the Developmental and Reproductive Toxicity (DART) study, female Wistar Han rats (44/group) were administered two 30 µg IM doses of BNT162b2 before mating and another two 30 µg doses during gestation (21 and 14 days prior to mating and on Gestation Days 9 and 20) (Fig. 3b). Half the females in each group underwent a cesarean section on Gestation Day 21, while the remaining females were allowed to deliver their offspring. Mothers and offspring were observed for 21 days after parturition. The vaccine was tolerated, and there were no vaccine-related effects on female fertility, pregnancy, or embryo-fetal or offspring development (Bowman et al., 2021). Anti-spike neutralizing antibody responses were present in the maternal females and their embryos and pups.
As toxicology data from the first repeat-dose toxicity study became available, it was shared with the US Food and Drug Administration (FDA) on a rolling basis, enabling the initiation of clinical trials 1 month after the Good Laboratory Practice (GLP) toxicity study was initiated. This streamlined process exemplifies the power of collaboration and the potential of modern molecular technologies in accelerating the development of life-saving vaccines (Fig. 4).

Timeline of BNT162 vaccine development compared with a traditional vaccine development timeline. Prior to the COVID-19 pandemic, most infectious disease vaccines took about 10 years to develop. Based on the previous research conducted, flexibility of the RNA modality, conduct of many activities in parallel vs sequentially, at-risk investments, and collaboration with health authorities, Phase 1 clinical development was begun within 1.5 months and emergency use authorization, conditional approval, or exception approval of BNT162b2 vaccine was achieved within 1 year of initiation of the first repeat-dose toxicity study.
The initiation of clinical trials marked a significant milestone in developing the COVID-19 vaccine, BNT162b2. Of the 5 vaccine candidates advanced into clinical trials, only BNT162b2 was selected to progress to Phase 3. This phase included over 44,000 subjects aged 16 and above and over 2,200 subjects aged 12 to 15. The vaccine demonstrated an efficacy of 91% against COVID-19 and almost 97% against severe disease. The vaccine was well-tolerated in clinical settings, with the most common reports being pain at the injection site, fatigue, and headache (Polack et al., 2020; Walsh et al., 2020; Thomas et al., 2021).
Post-market safety monitoring identified ADRs, including myocarditis and pericarditis (Comirnaty [package insert]). Comparing the nonclinical to the clinical data, there appeared to be strong translatability regarding neutralizing antibody and Th1 responses and local and systemic reactogenicity, with no safety risks identified for pregnant women or their infants/fetuses (Bowman et al., 2021; Sadarangani et al., 2022; Shimabukuro et al., 2021; Ciapponi et al., 2023; Lam et al., 2023).
Persistent demand for COVID-19 treatmentsTo combat the COVID-19 pandemic, a simultaneous two-pronged drug development strategy consisting of research directed to prophylactic vaccination and therapeutic treatments was adopted. Despite the vaccine’s success, there remained an urgent need for therapeutics to treat patients with low or no pre-existing immunity to circulating strains of SARS-CoV-2. In the early days of the pandemic, efforts focused on repurposing already licensed drugs, agents with existing nonclinical toxicology data, immunotherapies, convalescent plasma, and monoclonal antibodies or monoclonal antibody cocktails against the Wuhan variant spike protein. Antivirals for SARS-CoV-2, such as protease and S-protein inhibitors, were also developed.
The SARS-CoV-2 main protease (Mpro) is a virally encoded enzyme essential for viral replication. Mpro digests the virus p1a and p1ab polyproteins at multiple junctions to generate a series of proteins critical for virus replication and transcription, including RNA-dependent RNA polymerase (RdRp), the helicase, and Mpro itself (Fig. 5). The functional importance in virus replication, together with the absence of closely related homologs in humans, makes Mpro an attractive target for antiviral drug development.

Target of nirmatrelvir (PF-07321332) and lufotrelvir (PF 07304814) in SARS-CoV-2 lifecycle. SARS-CoV-2 infects cells through the ACE2 receptors with the lung and bronchial epithelial cells as the primary sites of infection. The SARS-CoV-2 Main protease is a virally encoded enzyme which is essential for viral replication. Main protease (Mpro) digests the virus P1a and P1ab polyproteins at multiple junctions to generate a series of proteins critical for virus replication and transcription. In vitro biochemical and cellular studies suggest that Mpro inhibitors nirmatrelvir (PF-07321332) and lufotrelvir (PF 07304814) interrupt the viral replication cycle through disruption of Mpro activity.
At Pfizer, we focused on advancing two protease inhibitors targeting Mpro, lufotrelvir (PF-07304814), and nirmatrelvir, using intravenous (IV) and oral routes, respectively (Fig. 6). Lufotrelvir (PF-07304814) is a phosphate prodrug of PF-00835231 (active moiety), which is a potent and selective inhibitor of the SARS-CoV-2 Mpro (Boras et al., 2021). Lufotrelvir (PF-07304814) has been investigated as a continuous IV infusion for treating patients hospitalized with COVID-19 (Robinson et al., 2023) .

Chemical structures of nirmatrelvir (PF-07321332) and lufotrelvir (PF-07304814).
The nonclinical safety profile was evaluated in vitro and in vivo systems (Boras et al., 2021; Robinson et al., 2023). Lufotrelvir (PF-07304814) and PF-00835231 were negative in the in vitro bacterial reverse mutation assay and did not induce micronuclei formation in vitro or in vivo. Both compounds had minimal potential for secondary (off-target) pharmacology at clinically relevant exposures and were compatible with human blood. The toxicity profile of lufotrelvir (PF-07304814) or PF-00835231 was assessed in GLP continuous IV infusion studies for up to 14 days in rats and cynomolgus monkeys. Continuous IV infusion was selected for in vivo studies since it was the intended route of clinical administration. The highest dose was selected based on the limit dose of 1000 mg/kg (for lufotrelvir [PF-07304814]) and the maximum feasible dose based on solubility (for PF-00835231). To enable rapid entry into clinical trials, single-dose, 24-hour continuous IV infusion, single-species toxicity with a 13-day recovery phase studies were conducted with lufotrelvir (PF-07304814) and PF-00835231 in rats.
This was followed up with 14-day continuous IV infusion toxicity studies in rats and monkeys to support clinical administration for up to 2 weeks. Lufotrelvir (PF-07304814) and PF-00835231 were tolerated and without test article-related effects in rats up to 1000 mg/kg. The infusion studies were associated with increased inflammation, also noted in the concurrent control groups in both rat and monkey studies; however, exacerbation of infusion procedure-related inflammatory effects by lufotrelvir (PF-07304814) was observed in the 14-day GLP monkey study, and the highest dose tested (1000 mg/kg/day) was considered above the maximum tolerable dose (MTD) (Table 3).
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/49_79-t3.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
These effects included changes in peripheral blood white blood cells (WBCs), coagulation parameters, inflammatory cell infiltrates in various tissues, and thrombo-emboli at the infusion site and/or in different tissues. The inflammatory effects are monitorable through peripheral blood chemistry, coagulation, and hematology evaluations. Another atypical strategy that we adopted to accelerate nonclinical studies was to use telemetered monkeys for the 14-day monkey study to concurrently conduct safety pharmacology risk assessments. There were no lufotrelvir (PF-07304814)-related quantitative or qualitative changes in any of the measured electrocardiograms, hemodynamics, or body temperature at doses up to 1000 mg/kg/day.
The first-in-human study was unusual because the first clinical administration was in hospitalized patients with COVID-19 rather than healthy volunteers (Robinson et al., 2023). This was done due to the urgent medical need for treatment. At the time of study initiation, single-dose rat toxicology data were available to support single ascending dose (SAD) administration in patients. Subsequently, data from the 2-week continuous infusion GLP toxicology studies in rats and monkeys became available to support the enrollment of multiple ascending dose (MAD) participants.
A study in healthy volunteers was also initiated that provided data to inform a starting MAD dose. The findings from the monkey study (exacerbation of infusion procedure‒related inflammation and thromboembolic) informed the pharmacokinetics (PK) stopping limits following consultation with the US FDA in the middle of conducting the SAD study. Continuous 24-hr and 120-hr IV infusions of lufotrelvir (PF-07304814) generally appeared safe and well-tolerated in this limited investigation in hospitalized patients with COVID-19 (Robinson et al., 2022).
Safety assessment of nirmatrelvir (PF-07321332)Nirmatrelvir (PF-07321332) binds to the active site of SARS-CoV-2 Mpro and forms a covalent interaction with the Mpro, as determined by the co-crystal structure. The binding mode of nirmatrelvir mimics the substrate binding, making numerous interactions with the protein that is analogous to enzyme-substrate contacts (Owen et al., 2021).
Nirmatrelvir (PF-07321332) demonstrated in vitro selectivity for coronavirus Mpro, showing little or no activity against a panel of human proteases, as well as HIV protease. In a mouse model, drug exposures reached severalfold above the 90% maximal effective concentration (EC90) and resulted in reductions in viral titers. Pathology assessments showed significant reductions in lung histopathology scores of alveolar thickening and perivascular infiltrates compared with vehicle controls. There was a dose-dependent decrease in replicating virus (Owen et al., 2021).
The nonclinical safety package for nirmatrelvir (PF-07321332) to support clinical development was consistent with the International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) M3 and included up to 4-week repeat-dose toxicology studies in rats and monkeys, a full genetic toxicology battery, full safety pharmacology battery, and assessments of DART (Sathish et al., 2022). There were no adverse findings in any of the repeat-dose GLP toxicity studies. The no-observed-adverse-effect-levels (NOAELs) were the highest dose administered in each study (Table 4). In rats, nonadverse, monitorable, and reversible clinical pathology findings included those suggestive of low-grade inflammation or alterations in the coagulation pathways without clinical or microscopic correlates. In monkeys, nonadverse, monitorable, and generally reversible clinical pathology findings included increases in alanine aminotransferase and/or aspartate aminotransferase and increases in fibrinogen at the high dose in the 1-month study without clinical or microscopic correlates.
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/49_79-t4.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
Nirmatrelvir (PF-07321332) had no adverse effects on male or female fertility in rats, fetal morphology or embryo-fetal viability in rats and rabbits, or pre- and postnatal development (PPND) in rats when evaluated at doses up to 1000 mg/kg/day. In the rat fertility and embryo-fetal development (EFD) studies, there were no adverse effects from nirmatrelvir (Catlin et al., 2022). In the rabbit EFD study, there were no nirmatrelvir (PF-07321332)-related effects on fetal morphology or embryo-fetal viability, although adverse nirmatrelvir (PF-07321332)-related lower fetal body weights were observed at 1000 mg/kg/day in the presence of low magnitude effects on maternal body weight change and food consumption at this dose. In the PPND study, there were no adverse effects of nirmatrelvir (PF-07321332) on the F0 (maternal/paternal) and F1 (first) generation.
Nirmatrelvir (PF-07321332) was not mutagenic or clastogenic in vitro genetic toxicity studies and was negative in the in vivo rat micronucleus assay incorporated into the 14-day GLP repeat-dose rat toxicity study.
Nonclinical safety assessments supported the clinical development of nirmatrelvir (PF-07321332). Nirmatrelvir (PF-07321332) has shown potent in vitro antiviral activity against all SARS-CoV-2 variants, including currently circulating Omicron XBB variants.
Collaboration with regulatory agencies underpinning the rapid development of vaccines and therapeuticsClose cooperation and collaboration with health authorities was a central factor that enabled the rapid development of COVID-19 vaccines and therapeutics. Worldwide, health authorities have facilitated speedy progress through several means, including timely meetings, scientific advice, removal of bureaucratic hurdles, acceptance of interim reports, and rolling submissions. In addition, several health authorities from various countries or regions developed specific guidance or reflection papers to help developers understand nonclinical requirements for pre-investigational new drug application (IND) meetings, new clinical trial applications (CTA), and IND, as well as authorization or approval. A select few are outlined in Table 5. Some of these guidances are no longer in effect since the end of the public health emergency.
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/49_79-t5.jpg%22>');%0A%09%09%09newWindow.document.close();%0A%09%09) Ensuring rapid, safe, and effective development of vaccines and therapeutics: lessons learned from COVID-19 for future pandemics
Ensuring rapid, safe, and effective development of vaccines and therapeutics: lessons learned from COVID-19 for future pandemics
Tremendous progress has been made in pharmaceutical R&D, and we were able to apply these learnings to accelerate drug discovery and development. Innovative development paradigms for COVID-19 vaccines and therapeutics during the pandemic, close collaborations with the health authorities, constant and timely communications at various milestones, and many development activities carried out in parallel (eg, chemistry, manufacturing, and controls (CMC), nonclinical, and clinical studies) all contributed to this success.
The COVID-19 pandemic has underscored the importance of swift and coordinated responses to global health crises. As we reflect on the lessons learned, several key strategies emerge that can guide future efforts in combating pandemics (Fig. 7).

Key lessons and emerging strategies from the COVID-19 vaccine and therapy development. Learnings from the COVID-19 vaccine and therapy development may be applied for future pandemic preparedness.
Overall, the lessons learned from the COVID-19 pandemic provide a strategic approach for potential future responses to global health crises. By fostering close collaborations, making at-risk investments, employing novel approaches, leveraging therapeutic expertise, and embracing innovative thinking, a rapid and effective response to future pandemics can hopefully be ensured.
Note related to animal study dataAll procedures performed on animals were in accordance with regulations and established guidelines and were reviewed and approved by an Institutional Animal Care and Use Committee or through an ethical review process.
Conflict of interestNasir Khan, Cynthia Rohde, and Jean Sathish are employees and/or shareholders of Pfizer, Inc.